The Investigation Gap: Why 48% of FDA Warning Letters Cite the Same Problem
Table of Contents
We analyzed 417 FDA Warning Letters issued to pharmaceutical and medical device manufacturers between January 2022 and January 2025. The analysis points to a clear conclusion: the binding constraint in manufacturing quality has shifted from detection to diagnosis, the cognitive work of determining why a defect occurred and whether the fix actually addresses it.
48% cite inadequate root cause investigation. The violations focus less on the defect itself and more on the investigation process. These deficiencies cluster into four failure modes: no investigation conducted, inadequate investigation, unidentified root cause, and CAPA that missed the root cause.
API, Oral Solid Dosage, and Medical device manufacturers are most frequently cited for RCA-related failures. All data and methodology for this analysis are open-sourced on GitHub.
The Investigation Gap
If you want to predict which manufacturers will receive an FDA Warning Letter, don't look at their contamination rates or their equipment age. Look at how they investigate problems.
The FDA publishes a convenient audit trail of where manufacturing quality programs fail: the Warning Letters database. We spent this past weekend reading through several of them, and a pattern emerged that we didn't expect: the citations aren't primarily about contamination, equipment failures, or process deviations. They're about how companies investigate those problems. With that, we committed to a thorough analysis of the database.
Scope and Methodology
Data source: FDA Warning Letters database.
Filtering logic: We focused specifically on CGMP violations (Current Good Manufacturing Practice) because these regulations explicitly mandate root cause investigation and corrective action procedures (e.g., 21 CFR 211.192 for pharma and 21 CFR 820.100 for devices). This makes CGMP the appropriate lens for assessing investigation capability.
By selecting for issuing offices that handle pharmaceuticals, medical devices, and biologics, we implicitly excluded several categories that operate under different regulatory frameworks or failure modes:
- Food, cosmetics, and tobacco: These involve different regulatory standards where RCA cited in a manufacturing context is less relevant.
- Clinical trial violations (GCP): These focus on clinical protocol adherence rather than manufacturing quality.
- Import alerts and labeling-only violations: These are often procedural or administrative rather than investigation-related.
- CDRH-only (Center for Devices and Radiological Health) letters without CGMP deficiencies: Focus on 510(k) or PMA (Premarket Approval) issues rather than systemic manufacturing quality.
Method: We classified letter text against four failure mode categories using regex patterns, validating the results with manual spot-checks. We also used an LLM (Gemma 3 4B) for product type classification and to extract defect types and claimed causes on our corpus.
Limitations: Keyword matching isn't perfect, and the dataset covers a specific 3-year window (January 2022 to January 2025). The data and methodology behind this analysis are open-sourced. You can find the full analysis and the underlying Jupyter notebook here: github.com/auxiliary-machines/fda-warning-letter-analysis.
Why This Matters: The Cost of a Warning Letter
Before presenting the findings, it's worth establishing the stakes. A Warning Letter is not a private conversation but a public accusation of federal law violations.
Public disclosure. Warning letters are posted on the FDA’s website within days and are generally available to customers, competitors, and investors. The market impact can be immediate: after Medtronic received an FDA Warning Letter in 2021, its stock fell more than 9%, leading to analyst downgrades from major firms like Wells Fargo and J.P. Morgan. Simultaneously, its direct rival, Tandem Diabetes Care, saw a 10.5% stock increase.
Immediate operational impact. The clock starts on receipt: companies have 15 working days to submit a comprehensive response. It must detail specific corrective actions for every observation, supported by evidence like updated SOPs and validation reports. Miss this window or submit an inadequate plan, and the FDA accelerates escalation—potentially seizing products, blocking new approvals (NDAs, 510(k)s), imposing import alerts, or filing injunctions to stop business operations entirely.
Operational and Industry Costs. Beyond individual stock hits, the systemic cost of regulatory failure is staggering. According to research published in the Journal of Industrial Engineering and Management, quality events—including Warning Letters, Form 483s, and consent decrees—cost the industry between 5 billion per year on average, with an additional 2 billion in lost sales. As far as we can tell, these estimates don't even account for the associated loss in market capitalization.
Escalation risk. Warning letters are a waypoint. Inadequate responses can lead to consent decrees—court-supervised agreements that can include production shutdowns, third-party oversight, daily fines (100 million fine to resolve a 1999 decree. (This decree lasted over a decade.)
When 48% of these letters cite investigation failures, better investigation capability isn't just a quality improvement but heavy-duty regulatory risk mitigation.
Finding 1: The Headline Number
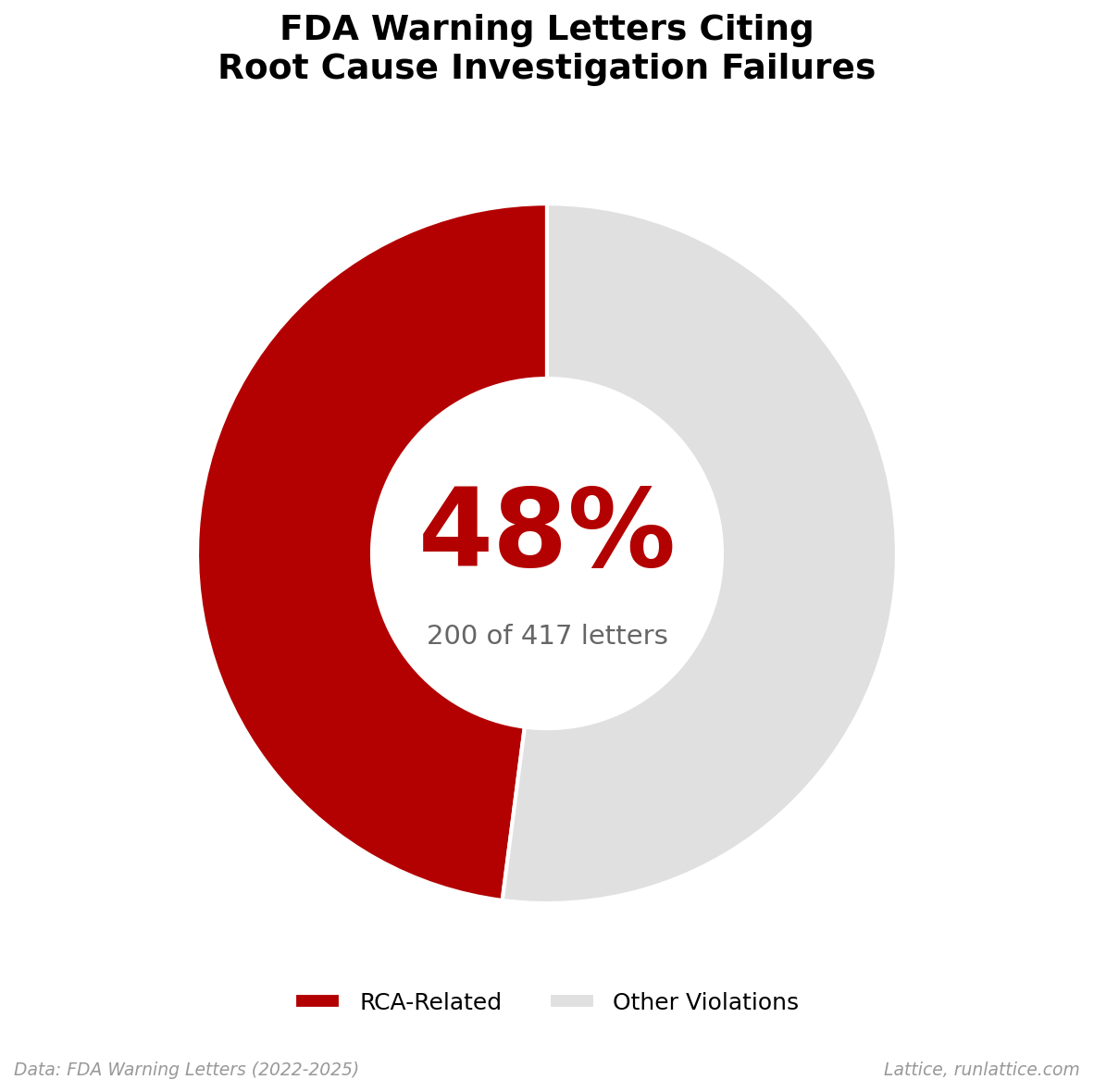
48% of warning letters cite root cause investigation failures.
These letters aren’t primarily about contamination events or equipment breakdowns, which are merely downstream symptoms. The FDA is citing the investigation process itself: companies that detected a problem, conducted an investigation, and arrived at a conclusion that the FDA deemed inadequate.
The pattern is consistent: investigations that stopped too soon, looked too narrowly, or proposed corrective actions that didn't address the actual cause. Nor is this a recent anomaly. When we analyze the data over time, the signal remains persistent.
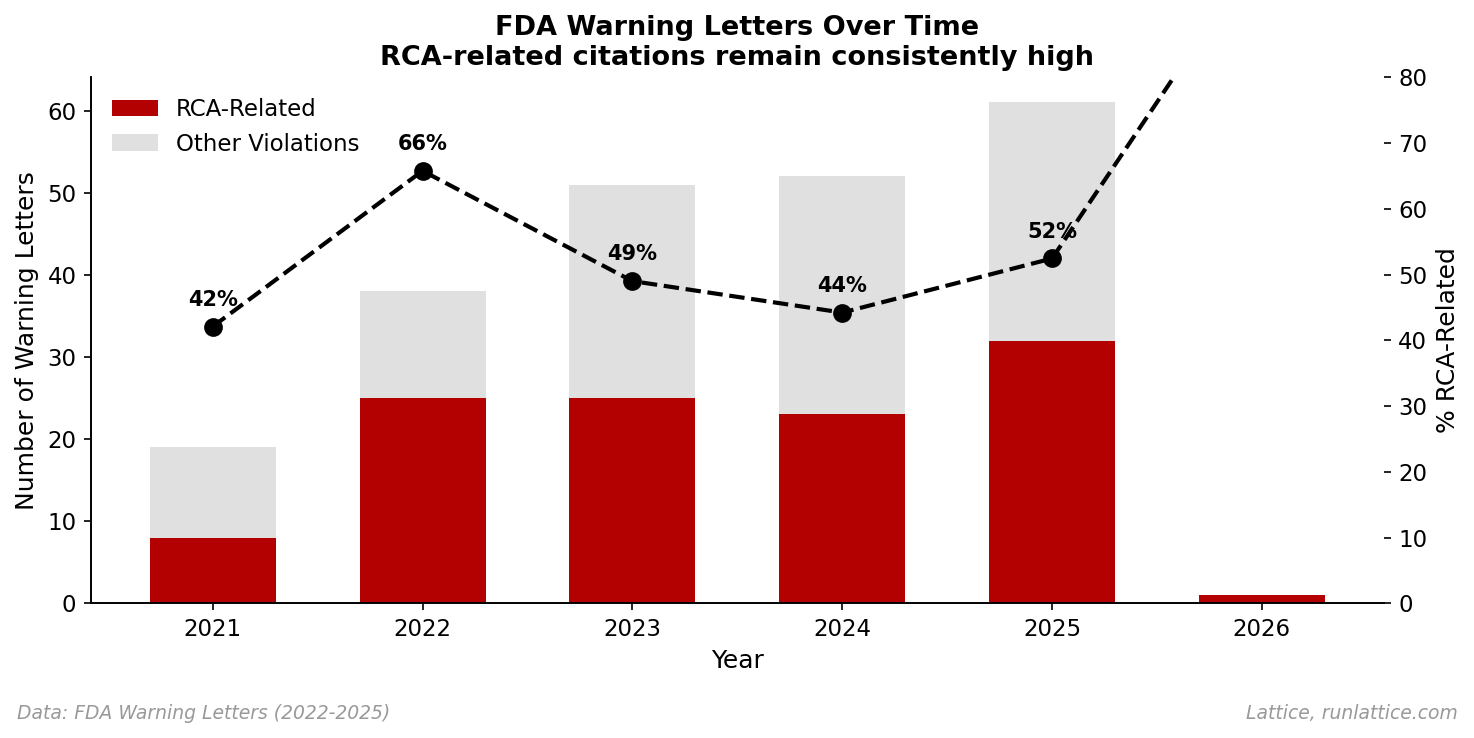
Despite the industry's focus on "Quality 4.0" and digital transformation, the fundamental capability of diagnosis hasn't improved. If anything, the complexity of modern manufacturing processes has outpaced our ability to investigate them.
Finding 2: Where Investigation Breaks Down
The deficiencies cluster into four categories. Note that these overlap; a single letter often cites multiple failures, reflecting compound breakdowns in the process.
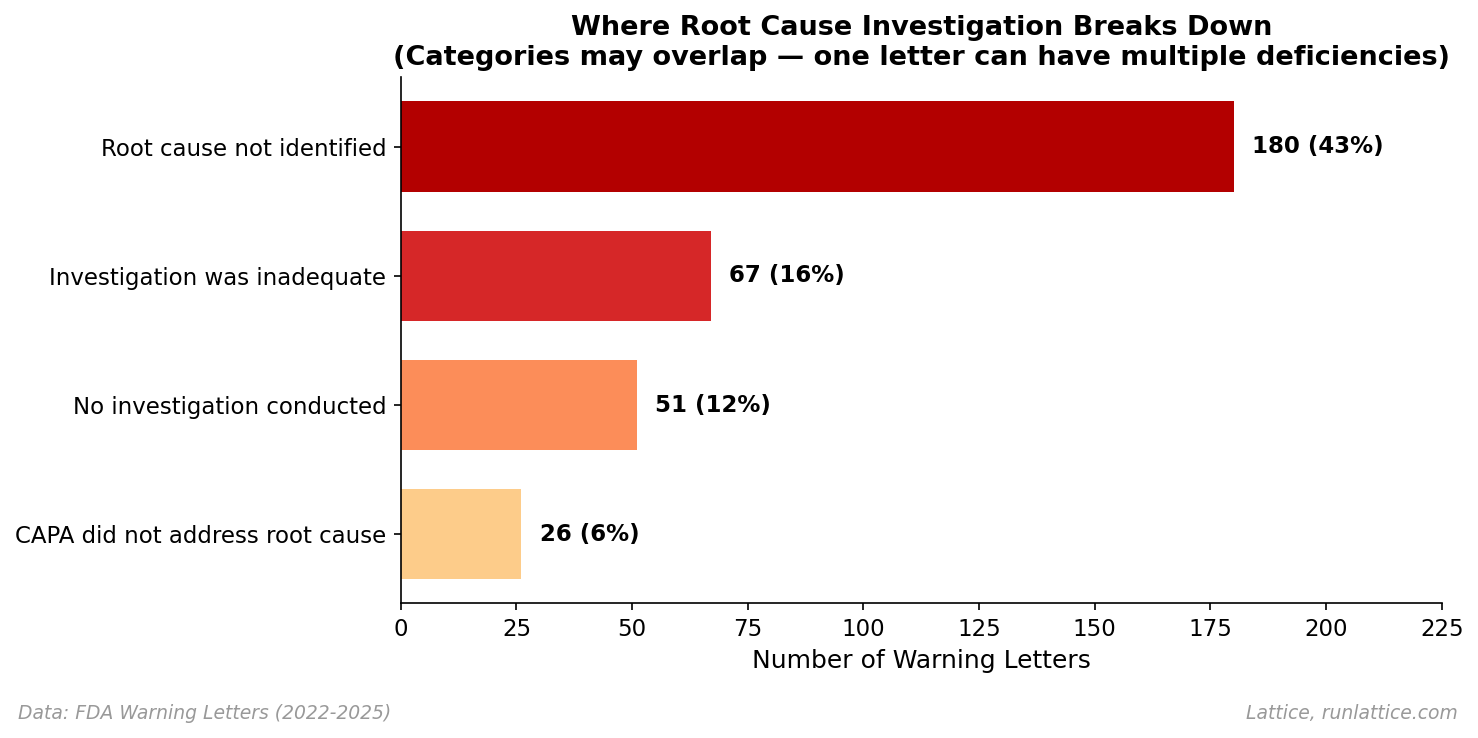
- Root cause not identified (43%). The company investigated but stopped short of determining the underlying cause. The FDA’s critique often highlights that the investigation identified what happened but not why. A common pattern is attributing failures to "operator error" without explaining what conditions allowed the error to occur.
- Investigation was inadequate (16%). The investigation was superficial, incomplete, or methodologically flawed. The FDA frequently cites a failure to consider all potential sources, a failure to extend the investigation to other potentially affected batches, or a lack of scientific justification for conclusions.
- No investigation conducted (12%). The company simply didn't investigate. This often occurs with out-of-specification (OOS) results that were retested and released without investigating the initial failure.
- CAPA did not address root cause (6%). The corrective action missed the mark. The company identified a cause (or claimed to), implemented a fix, but the fix didn't actually address the underlying issue. The FDA critiques these as treatments of symptoms rather than causes.
The most common failure is also the most subtle: investigations that technically occurred but either didn't go deep enough or were documented/reported superficially–that latter is what we call the "lossy compression" problem.
Finding 2.1: The Compounding Effect
It’s rarely just one thing. When we looked at the complexity of these citations, we found that 45.5% of RCA-related letters cite multiple failure modes, with violations often coming in pairs or triplets.
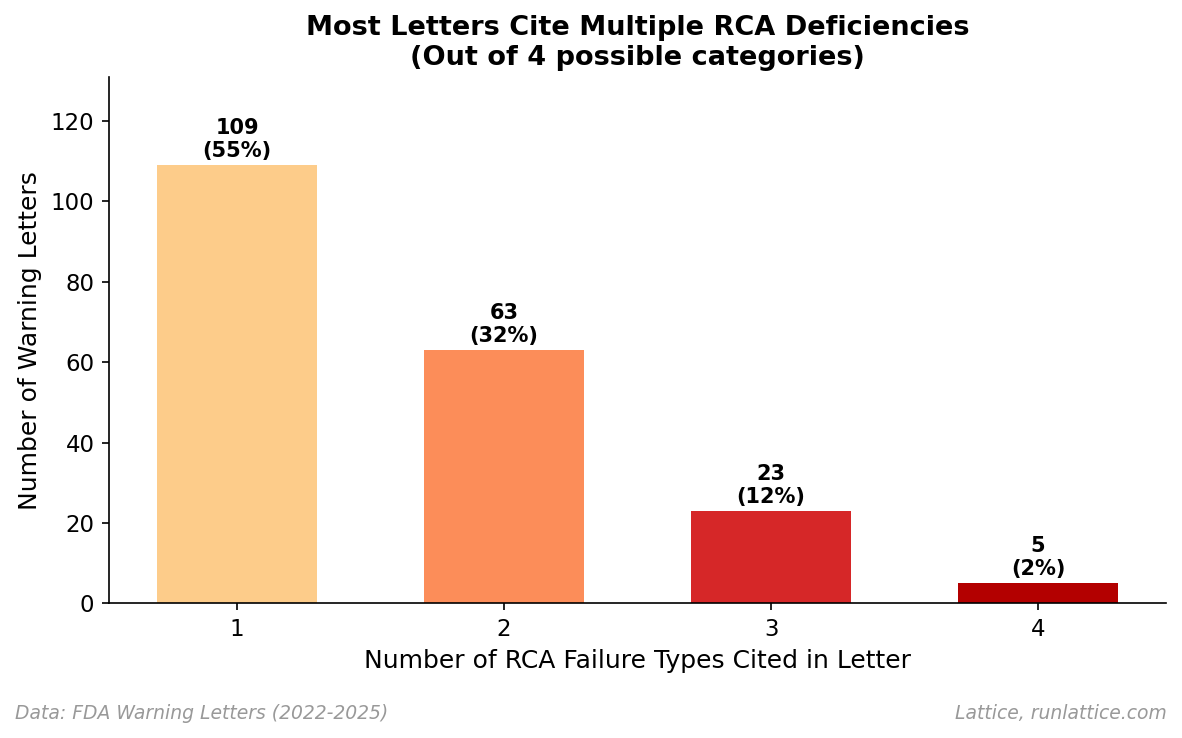
This suggests that when an investigation process is broken, it is often broken at multiple levels. An organization that lacks the cognitive tools to identify the true root cause (Category 1) is frequently the same one that lacks the rigor to define the investigation scope correctly (Category 2) or implement a CAPA that actually fixes the underlying problem (Category 4). These aren't isolated errors as much as symptoms of a fragility in the entire reasoning capability of the organization.
Finding 3: Who Gets Cited
Breaking down the 200 RCA-related letters by product type shows that investigation failures are a cross-cutting capability gap, though some sectors face more scrutiny.
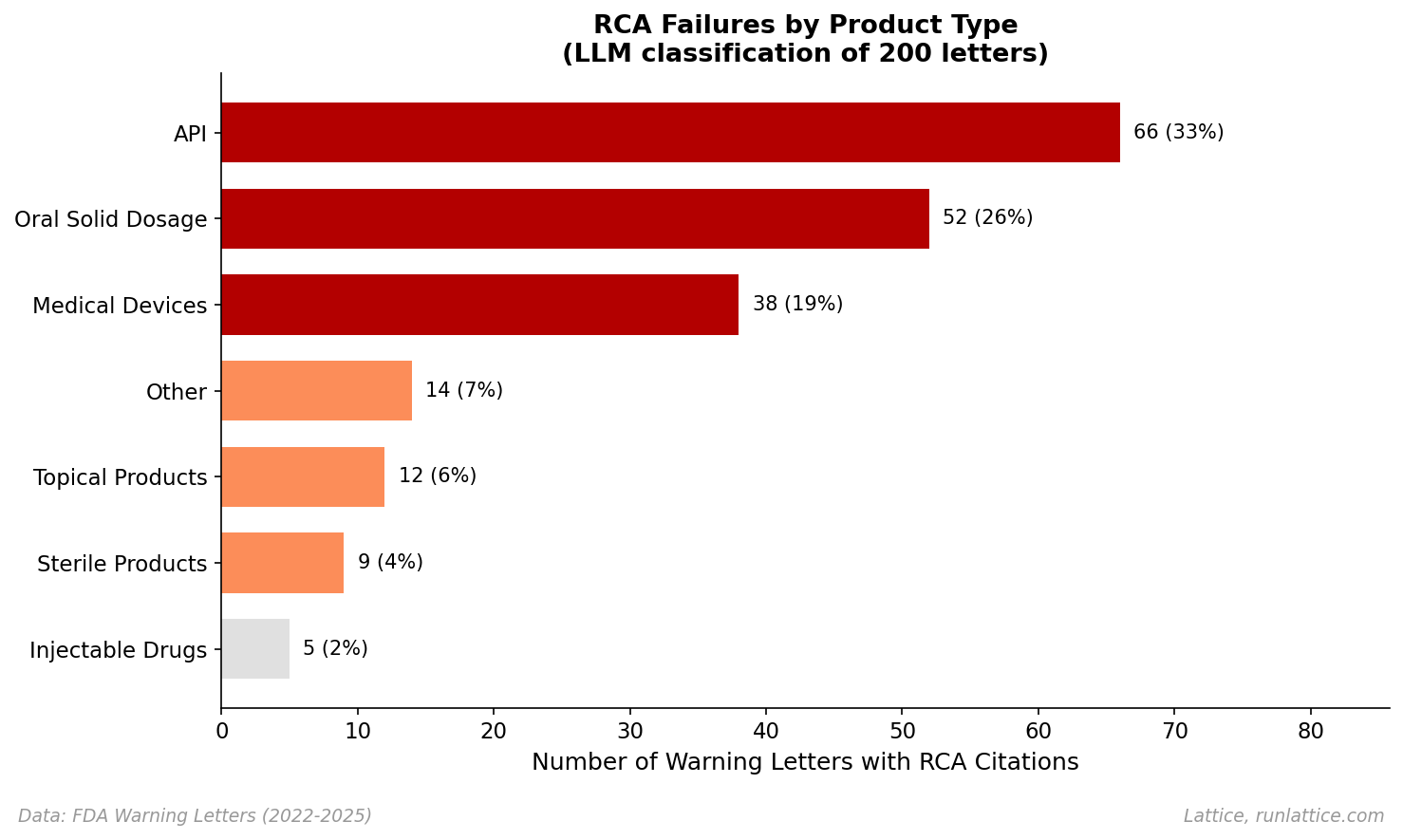
API manufacturers account for the highest absolute count (33%), followed by Oral Solid Dosage (26%). Medical devices represent 19% of the RCA-related citations (38 letters). Device manufacturers face QSR (Quality System Regulation) requirements with explicit CAPA mandates, leading to frequent citations when those processes fail. Sterile and Injectable drugs represent about 6% combined, where the stakes of contamination are highest.
While this distribution reflects inspection frequency and regulatory focus, it confirms that no manufacturing sector is immune to the "investigation gap."
Examples: What "Inadequate" Looks Like
Let's look at five specific examples from our dataset. They illustrate the diverse ways investigations fail to satisfy regulatory standards. These failures generally follow four recurring patterns:
- "Human Error": Attributing a defect to operator error (like "pipetting mistakes") without scientific proof or investigating the conditions that allowed the error to occur.
- Artificial Narrowing: Limiting the scope of an investigation to a single batch or a single component, even when evidence suggests a systemic risk to other products or materials.
- Silent Failures: Closing an investigation simply because a physical sample wasn't returned or a test result couldn't be instantly replicated, rather than investigating the process history or library samples.
- Narrative Compression: Treating systemic issues—like a 20% failure rate or recurring data integrity gaps—as isolated, one-off deviations rather than a loss of process control.
| Company & Reference | Defect | Claimed Root Cause | FDA Finding |
|---|---|---|---|
Professional Disposables International Inc. | OOS assay results | "Pipetting error" or "incorrect dilution" | Attributed failure to human error without evidence; failed to retain glassware or expand investigation to other batches. |
Accra-Pac Inc. (Voyant Beauty) | Benzene contamination | Limited to isobutane propellants | Artificially narrow scope; limited review to one propellant type despite evidence of wider contamination. |
Zyno Medical LLC | Leaking admin sets | "Device not returned; no cause established" | Failed to investigate because unit wasn't returned; did not review manufacturing records or test retained samples. |
Missouri Analytical Laboratories | Falsified autoclave records | "Known laboratory errors" | Treated systemic data falsification as a simple error; failed to assess extent of integrity breaches across operations. |
Sanofi (Genzyme Corp) | 20% bioreactor failure rate | Varied individual deviations | Treated massive systemic failure rate (1 in 5 runs rejected) as isolated incidents rather than a fundamental loss of process control. |
Investigations as Lossy Compression
The pattern across these letters is intellectually consistent: companies detect a problem, conduct an investigation, and arrive at a conclusion. But the investigation is lossy. It compresses away the interaction effects, the temporal correlations, and the informal adaptations that actually explain why the defect occurred.
This maps to our concept of adherence effort: the true effort required to keep a system operating within its designed constraints. Every manufacturing process accumulates drift, including small deviations, workarounds, and compensations that maintain output despite degraded conditions. These adaptations form an invisible economy that keeps the system running, but they also obscure true causation.
When a defect occurs, the investigation must navigate this invisible economy to find the actual failure mode. Most investigations don't. They identify a proximate trigger ("operator didn't follow procedure") without asking why the procedure was bypassed, what conditions made the bypass seem reasonable, or whether the same conditions exist elsewhere.
The result is narrative compression: the investigation produces a plausible story that satisfies the immediate need for closure, but discards the systemic information needed to actually prevent recurrence.
These failures stem from methodology and tooling gaps rather than a lack of diligence. Investigations are cognitively demanding. They require synthesizing data across systems, correlating events across time, and reasoning about interactions that aren't obvious from any single data source. Most quality teams do this manually, under time pressure, with spreadsheets and tribal knowledge. The 48% figure isn't surprising when you understand the gravity of the task.
Implications
What does this mean for quality teams? Three takeaways stand out.
Detection is necessary but not sufficient. You can have world-class inspection and still fail investigations. The FDA cites companies less for finding defects and more for failing to understand them.
Investigation is a cognitive bottleneck. Root cause investigation is fundamentally a reasoning challenge rather than a labor constraint. Adding more people to an investigation often adds confusion rather than clarity. It requires tools that can survive the synthesis of disparate data points.
The cost of inadequate investigation is compounding. You don't just fail the current investigation; you fail to learn, allowing the same failure mode to recur. The FDA sees these patterns across inspections. The companies that avoid escalation are those that investigate effectively and demonstrate systemic improvement.
Some of these failures are discipline problems, where companies simply didn't investigate. But the majority are reasoning problems. These are investigations that occurred but didn't go deep enough, didn't extend far enough, or proposed fixes that didn't match causes. These are the failures that Lattice can address.
| Failure Mode | Primary Barrier | Lattice Relevance |
|---|---|---|
| No investigation conducted | Discipline / Culture | Low. This is a management enforcement issue. |
| Investigation was inadequate | Process / Rigor | High. Lattice workflows enforce completeness and standards. |
| Root cause not identified | Reasoning / Cognitive | High. Lattice assists in synthesis and diagnosis. |
| CAPA did not address root cause | Logic / Alignment | Medium. Lattice verification can flag logical mismatches. |
An effective investigation surfaces not just the proximate trigger but the contributing conditions: which process parameters drifted, which adaptations accumulated, which signals were present but unnoticed. It extends automatically to other potentially affected batches. It produces thorough documentation without manual reconstruction.
Closing
Fixing the investigation gap requires treating investigation as a first-class operational capability: systematic, tool-supported, and integrated into the quality workflow rather than bolted on after the fact.
At Lattice, we’re building the infrastructure to make this possible. By automating the evidence-gathering and reasoning process, we help teams move past "operator error" and find the systemic causes that actually matter.
Cite this Research
If you are referencing this data in your own publication, please use the following citation:
Auxiliary Machines Inc. (2026). The Investigation Gap: Why 48% of FDA Warning Letters Cite RCA Failures. https://www.runlattice.com/blog/the-investigation-gap
For media inquiries or data requests regarding the full corpus of 417 letters, contact [email protected].